
H-Optimus-1 for streamlining Spatial Transcriptomics workflows in IBD research
Executive Summary
An international research team at a top-ranking university, specializes in inflammatory bowel disease (IBD) and the underlying mechanisms of disease development. Their work harnesses multi-omics and spatial technologies with machine learning to identify pathogenic cell types and pathways, aiming to improve disease prediction and treatment.
With an aim to overcome the challenge of subtle and costly disease signals in IBD, the team used Bioptimus’s foundation model, H-Optimus-1, to predict spatial gene expression directly from routine H&E pathology slides. This allowed them to triage the most informative slides and regions for spatial transcriptomics (ST) assays revealing progression-driving niches and pathways. In evaluating several foundation models, H-Optimus-1 ranked among the top performers, providing accurate and explainable slide-level predictions.
This case study demonstrates how the H-Optimus-1 foundation model can streamline disease research, cutting costs and time by turning routine H&E slides into predictive maps, ensuring that only the most informative ST assays are run.

The Context: From Slides to Spatial Signatures—Understanding IBD in Situ
Inflammatory bowel disease (IBD) is a group of chronic, relapsing inflammatory disorders of the gastrointestinal tract driven by an inappropriate immune response in the gut. The two main forms of IBD are Crohn’s disease (CD) and Ulcerative colitis (UC). CD can affect any part of the GI tract (often terminal ileum and colon), with transmural inflammation leading to strictures, fistulas, and granulomas. UC is limited to the colon and rectum, with continuous mucosal inflammation causing bloody diarrhea and urgency.
IBD arises when a genetically primed immune system mounts a persistent, dysregulated response to gut microbes across a compromised epithelial barrier, shaped by environmental exposures and culminating in chronic inflammation and tissue remodeling. Its underlying mechanisms are difficult to understand because it is a heterogeneous disease with subtle morphology–molecular links. It is further complexified by its chronic and relapsing nature, lasting periods of up to several decades.
Many relevant molecular programs differ only subtly in H&E, and some signals are rare or patchy, making them hard to detect and generalize across tissues, stains, and scanners. Therefore, H&E slides and ST assays provide complementary information on morphology and molecular readouts, which can be combined to provide researchers more information about underlying disease mechanisms.
The Challenge: Current Limitations of Spatial Transcriptomics
ST technologies measure gene expression in tissues at resolutions that range from subcellular to micrometer‑scale, enabling maps of cell types, states, and neighborhoods. Researchers can use these spatial readouts to connect molecular programs to visible tissue features, form mechanistic hypotheses, and plan targeted follow‑up experiments.
Two widely adopted and commercially available ST platforms are Visium and Xenium by 10x Genomics, which are sequencing-based capture and imaging‑based targeted in-situ platforms, respectively. While both technologies in principle enable the measurement of molecular features in the spatial context of the tissue slide, they both come with limitations preventing discovery at scale.
Visium reads molecular features of the whole transcriptome using spatially barcoded spots at fixed physical positions allowing a scaled discovery with respect to the number of genes measured locally, however the capture area of a Visium slide is physically limited by a few mm, preventing scientists to measure large tumor sections and requiring them to select the most promising tissue area upfront. In contrast, Xenium (imaging‑based) uses lower-plexed, 200-5000 genes, targeted probe panels to detect RNAs at single‑transcript, subcellular resolution on FFPE or fresh‑frozen sections, offering a hypothesis driven approach to understand molecular and cellular differences in-situ. Xenium offers a slightly larger capture
area up to a few centimeters, however is more limited by the run-time and running unbiased large-scale whole transcriptome based experiments is to-date not practical.
The ST workflow is made up of the following steps (Figure 1):
● Tissue preparation (i.e. frozen, fixed, embedded, and sectioned)
● Sample mounting onto ST technology-tailored slide
● Sample processing by platform chemistry to produce spatially resolved transcriptomics gene‑expression profiles.
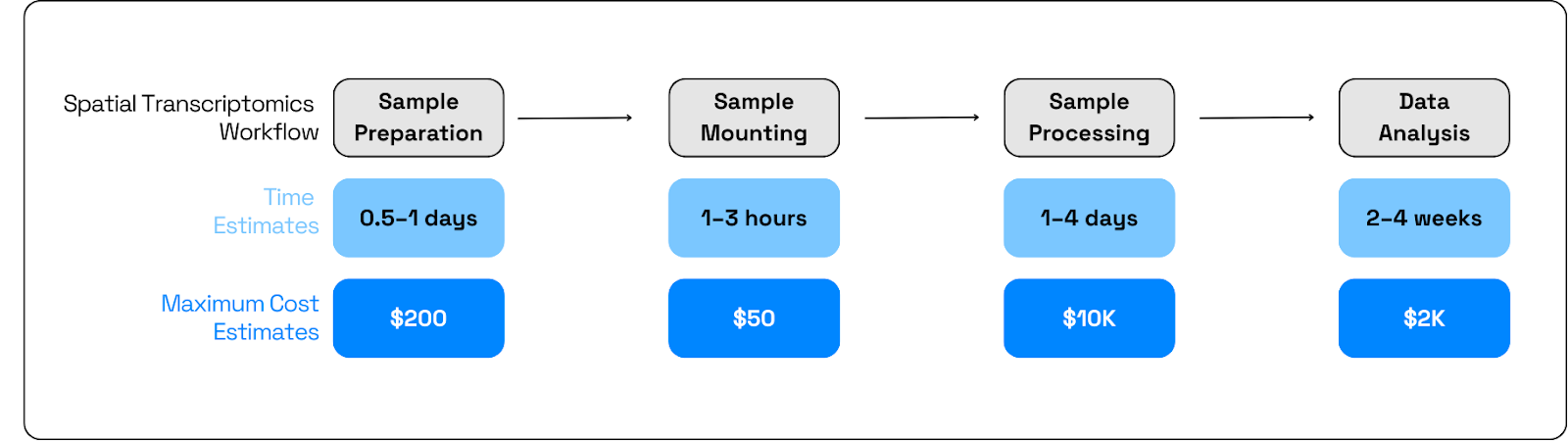
The use of spatial transcriptomics in cohort-scale studies is constrained by both time and cost, slowing hypothesis testing. It’s exacerbated by the chronic nature of the disease, which requires ST assays to be performed on historical tissue sections. Typically ST sample costs range in the thousands of dollars, depending on capture area and run format. Further limiting throughput and time are: imaging‑based platforms span from ~52 mm²/week to ~1,400 mm²/week, and practical wet‑lab plus analysis cycles often extend to multi‑day runs and weeks to interpretable results, several days of instrument time per slide before downstream processing and QC can begin. These constraints make optimizing the use of the highest value slides and identifying precise regions of interest critical.
In real‑world IBD cohorts, tissue composition varies widely across patients, sites, and sections; with many biologically important signals coming from rare cell types or transient states that are sparsely represented. Many models perform well on abundant epithelial programs, but miss early or rare drivers of disease. Therefore, predictors must generalize across various signals, ranging from global morphological features to patient-distinct effects that are often linked to shifts in cell-type abundance or cell phenotypes present in only a small subset of the cohort’s patients.. Additionally, models need to overcome the technical batch effects, such as incomplete staining,scanner variability, and differences in slide preprocessing batch to batch and from different facilities.. Practically, this demands diverse training data, careful validation across cohorts and sites, and evaluation metrics that weight both head (frequent) and tail (rare) biology so that insights extend beyond the obvious to the mechanistically meaningful.
The Solution: Reducing Spatial Assay Costs via Foundation Model–Driven Triage
To overcome these challenges, the team designed a slide-to-gene-expression prediction system incorporating a foundation model, which offers several key advantages. First, it enables smart spending by prioritizing specific slides and regions for costly Spatial Transcriptomics (ST) assays. Secondly, it provides access to historical cohorts, allowing for the longitudinal identification of early disease signals—a crucial factor in understanding a chronic condition. Finally, it generates robust, transferable representations that generalize across heterogeneous cohorts, including rare, clinically meaningful signals.

A foundation model (FM) is a large, pre‑trained AI model backbone that encodes rich morphology into generalizable embeddings. These are then used as inputs to a lightweight prediction head that is a small task‑specific model trained on paired H&E-ST data to map embeddings to genes or signatures. This combined approach outperforms training a task model from scratch because it starts from broad, data‑rich priors, needs less labeled data, resists overfitting to cohort quirks, and transfers across scanners and stains (Figure 3).
In this type of setting, H&E images are digitized and tiled; the FM produces patch‑level embeddings; lightweight heads predict gene/signature scores per patch; scores aggregate to slide‑ and ROI‑level maps to prioritize Xenium regions; measured spatial readouts are co‑registered to close the loop, refine heads, and update ROI criteria. The FM is the invariant representation layer that anchors the entire workflow.
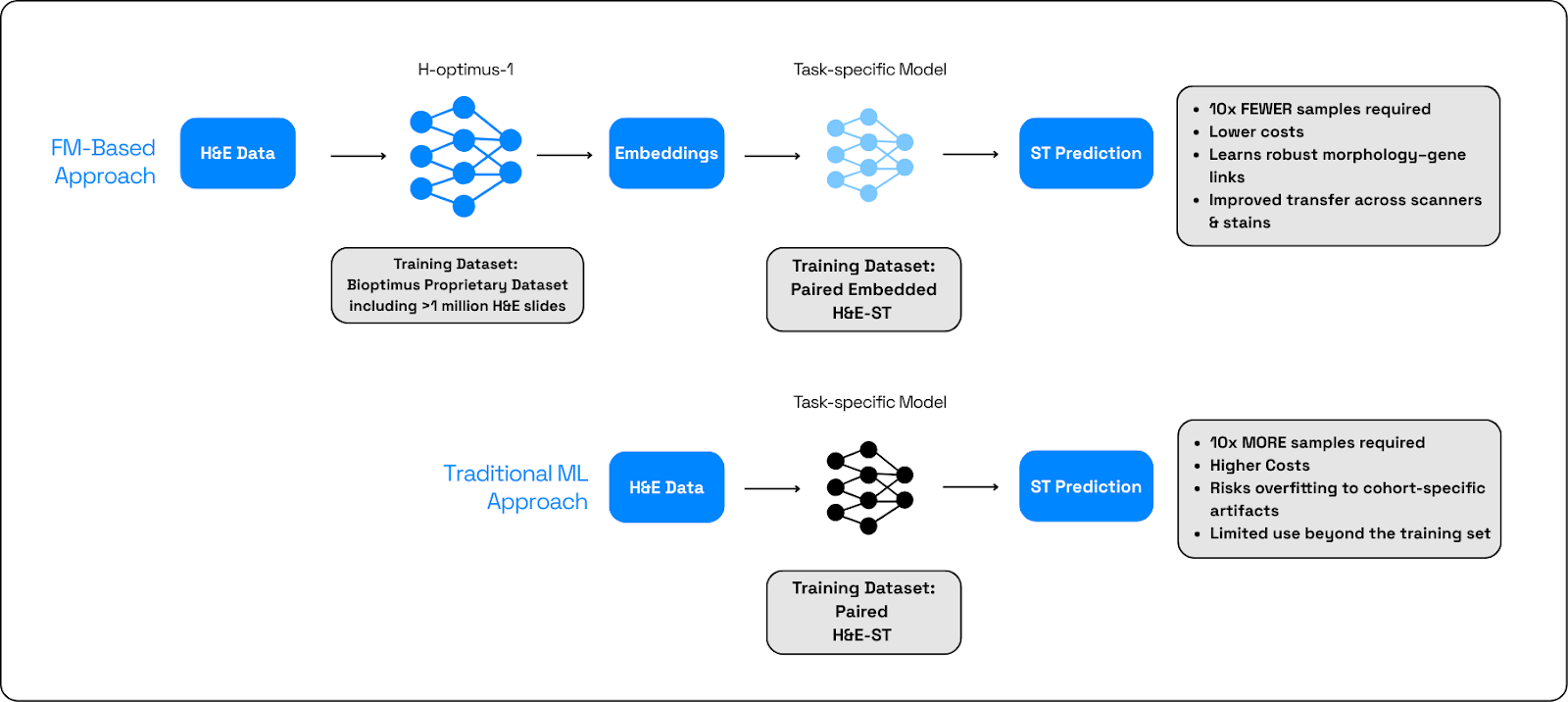
But not all FMs are made equal. Models trained on broader, more heterogeneous histology better encode morphology that is invariant across tissues, labs, stains, and scanners, which improves out‑of‑cohort generalization. Their embeddings also facilitate modality bridging: even when spatial panels differ (gene sets, platforms), the shared morphology space enables prediction heads to learn consistent mappings, capturing both common and rare signals while reducing sensitivity to limited sample sizes.
The research team compared 16 backbones, with the evaluation done on both the top 50 most variable genes and the entire gene panel, in a leave-one-patient-out cross validation manner. The results showed H‑Optimus‑1 to be a top performer for predicting spatial gene expression from H&E1. This can be partly attributed to H‑Optimus‑1 being trained on large, diverse slide collections, including substantial GI content, to learn high‑fidelity tissue representations that preserve cellular architecture and context—features critical for mapping morphology to spatial gene programs. It has been used successfully for histology‑to‑omics inference tasks and slide triage in research settings, making it a strong fit for H&E→ST prediction and ROI selection2.
This approach leverages a broad base of prior learning while remaining flexible to their IBD use cases and evolving assay setups.
The Impact
1. Foundation Models help cut costs and time of ST assays
The triage workflow, guided by H-Optimus-1, enables the team to select the most promising slides and regions for costly Xenium runs. This targeted approach significantly improves the efficiency of spatial data generation, ultimately accelerating the pace of discovery.
By using H&E-driven predictions to guide spatial assays, the team is able to reduce
experimental waste and increase the "hit rate" of informative experiments. This focus lowers the cost per discovery, making the research process more resource-efficient.
The rapid slide-level scoring capabilities of the system allow for faster iteration on experiments and hypotheses concerning inflammation signatures, niches, and biological responses. This ability to rapidly test and refine hypotheses before committing to expensive runs streamlines the research cycle and drives faster progress.
2. Foundation Models provide robust predictions when cohorts are small
The team's system demonstrates the power of foundation models, with H-Optimus-1 ranking among the best-performing models for predicting spatial gene expression signatures directly from H&E images, even with limited cohort sizes.
The team notes that H-Optimus-1 delivers robust performance on their current, relatively small dataset. However, reliably predicting rare cell types remains challenging. This finding highlights the need for additional data and future multi-omic integration to overcome current limitations.
This is precisely where the capabilities of our newest model, M-Optimus, can provide a significant step forward. M-Optimus is being trained on a massive, anonymized dataset comprising both whole-slide pathology images and corresponding multi-omics data, from diverse tissue types and disease states, with a particularly rich representation of GI pathology. This extensive and multimodal training will help M-Optimus learn finer-grained, subtle morphological features and their correlation with spatial molecular programs, which is crucial for reliably predicting rare cell states and early disease drivers, ultimately enhancing the robustness and clinical relevance of the slide-level triage.
3. Unlocking IBD Mechanisms to Improve Lives and Outcomes
Understanding the underlying mechanisms of IBD is an urgent need. Ulcerative colitis (UC) and Crohn's disease (CD) affect an estimated 6 to 8 million people worldwide, severely degrading quality of life, disrupting daily activities, and driving up healthcare costs. Despite available therapies, a significant number of patients face challenges: up to 30% do not respond initially, up to 50% lose response over time, and critical complications like intestinal fibrosis still lack effective antifibrotic treatments.
The team’s approach unlocks molecular insights from H&E scans over long timecourse studies,opening access to historical cohorts, identifying early signals of IBD complications. By uncovering how symbiotic microflora, intestinal epithelial cells, and the immune system interact to trigger and sustain inflammation and fibrogenesis, we can move beyond trial-and-error care toward precise, durable interventions. This knowledge allows for better matching of patients to effective, targeted therapies, preventing progression before irreversible damage occurs, and ultimately reduces surgeries, hospitalizations, and the day-to-day burden that keeps people from work, family life, and long-term wellbeing.
Biological interpretability, supported by spatially grounded predictions, further aids this effort by supporting hypothesis generation about why certain patients progress or recur, and which cell programs may drive disease states.
Conclusion
The research team demonstrates the powerful potential of H-Optimus-1 in overcoming limitations of cutting-edge ST assays. By predicting spatial gene expression directly from routine H&E slides, H-Optimus-1 enables a 'smart-spending' approach, which significantly reduces experimental waste, lowers the cost per discovery, and accelerates the pace of mechanistic research into complex, heterogeneous diseases like IBD. The model’s ability to provide robust, generalizable predictions,even in small, real-world cohorts, establishes a scalable framework for integrating morphology and molecular data.
Looking ahead, the success of this foundation model-driven slide triaging paves the way for further advancements. We will build on the work achievable with H-Optimus-1 using our upcoming multi-modal, multi-scale M-Optimus model, which will incorporate an impressive range of data modalities and promises to offer enhanced predictive power for rare and subtle disease drivers.
About H-Optimus-1
H-Optimus-1 is Bioptimus’s state-of-the-art foundation model for pathology, trained on large, diverse cohorts of whole slide images to learn generalizable tissue representations. The model has achieved strong results across standard benchmarks, including gene expression prediction and metastasis identification, and is being adopted across research and development workflows.
Bioptimus contribution
Bioptimus provides H-Optimus-1 access, model guidance, and technical collaboration to help researchers adapt foundation models to their biological questions, accelerating experimental design and mechanistic discovery in complex disease areas like IBD.
References
1. Bioptimus. H-Optimus-1, 2025. URL
https://huggingface.co/biOptimus/H-Optimus-1
2. Jaume G., Doucet P., Song A. H., Lu M. Y., Almagro-Perez C., et al. 2024. HEST‑1k: A Dataset for Spatial Transcriptomics and Histology Image Analysis. Advances in Neural Information Processing Systems. Link — Dataset card: HEST‑1k on Hugging Face