
From Fragments to Maps: M-Optimus Decodes the Molecular Geography of Cancer
TL;DR:
- 60% More Accurate: By looking at multiple biological languages at once, M1 delivers up to 60% better performance than well-established competing models.
- The Power of One General Model: We proved that one generalist brain is more powerful and versatile than many small, specialized models.
- Unlimited Growth: M1 gets smarter as it sees more data. Through our STELA program—an ambitious effort to build a global biological database—we are fueling M1 with the data it needs to master human biology.
- Mining the Past: Analyzing decades of historical clinical trials at a fraction of the original cost to find out why certain drugs worked for some but not for others.
- Better Target Discovery: Finding needles in haystack biomarkers to help get the right drugs to the right patients faster.
- Seeing the Invisible: Characterizing tumors with a level of detail that is literally invisible to the human eye, even under a microscope.
Biology has always been studied in fragments
- Complexity: Biology is so intricate that traditional computational tools simply couldn't process these layers holistically.
- Scarcity: Truly cross-modal data, where every layer is measured for the same patient at the same time, has been extraordinarily rare.
Introducing M-Optimus: one model for biology
- H&E Pathology (Hematoxylin & Eosin): the pathology slide image collected from a biopsy or during surgical resection that today represents the primary diagnostic tool at the bedside for physicians. The H&E shows the cellular morphology and architecture of the tumor.
- Bulk RNA-seq: A measure of total gene expression in a sample. This shows what genes that are expressed and the signaling pathways turned on in that tumor and within the organ surrounding the tumor.
- Spatial Transcriptomics: A high-definition portrait showing exactly where gene activity is happening. While bulk-RNA gives you an aggregate picture of the genes expressed, spatial transcriptomics shows which cell types are activated, how the patient’s adaptive immune system is reacting to the tumor, and how these cells are interacting at a molecular level with the surrounding tissue.
- The AI Invention. Solving this challenge required moving beyond the limitations of existing AI frameworks. Hence, we developed a proprietary model architecture and training methodology. This new blueprint is uniquely flexible. It converts a patient’s tissue data into a digital feature map that can natively predict the activity of 20,000 genes simultaneously. It can generate these insights from a standard H&E slide alone, or improve its accuracy by incorporating Bulk RNA data when available. This digital foundation can then be easily fine-tuned to predict almost any other property of a tumor.
- The Data Moat. To teach an AI to connect these dots, you need "ground truth" data where all three layers are measured on the same patient. While such comprehensive data is notoriously difficult to source, Bioptimus has bridged this gap through strategic partnerships. We trained M1 using a unique proprietary dataset comprising (i) millions of H&E whole slide images from more than 50 organ tissues, and (ii) thousands of patient records where H&E, bulk and spatial transcriptomics are paired and aligned.
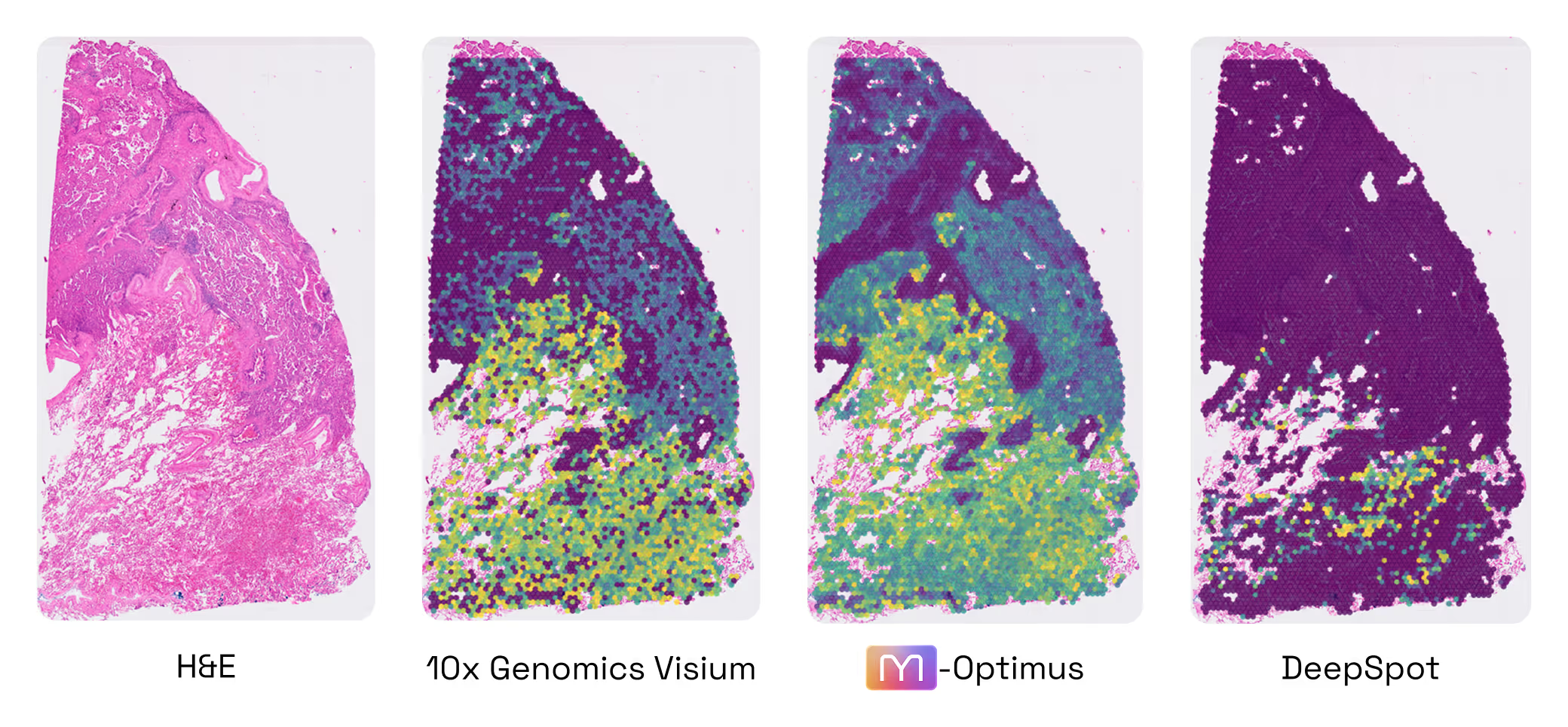
M1 performance: the benefits of multimodality

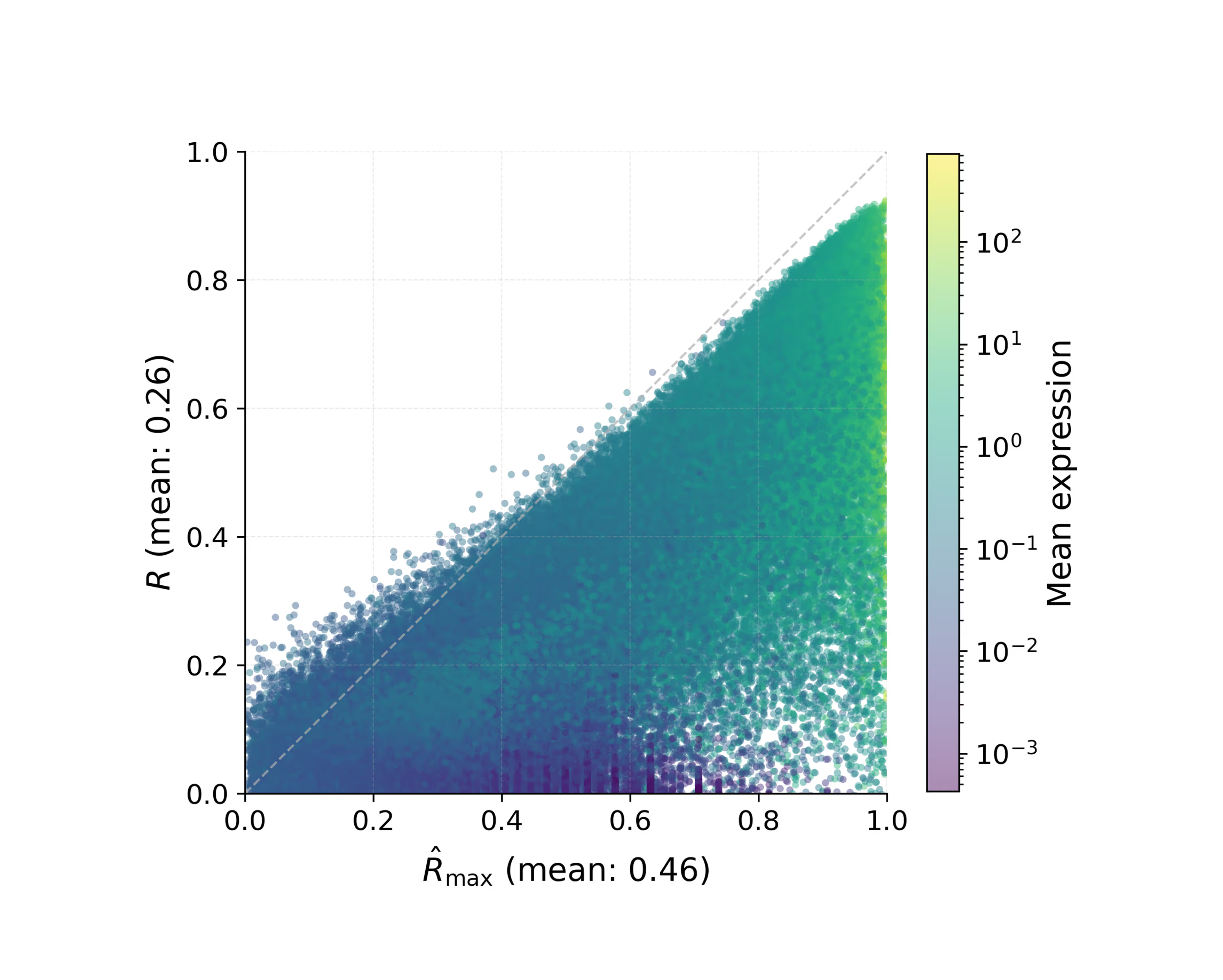
- Predict SpTx from H&E without fine-tuning
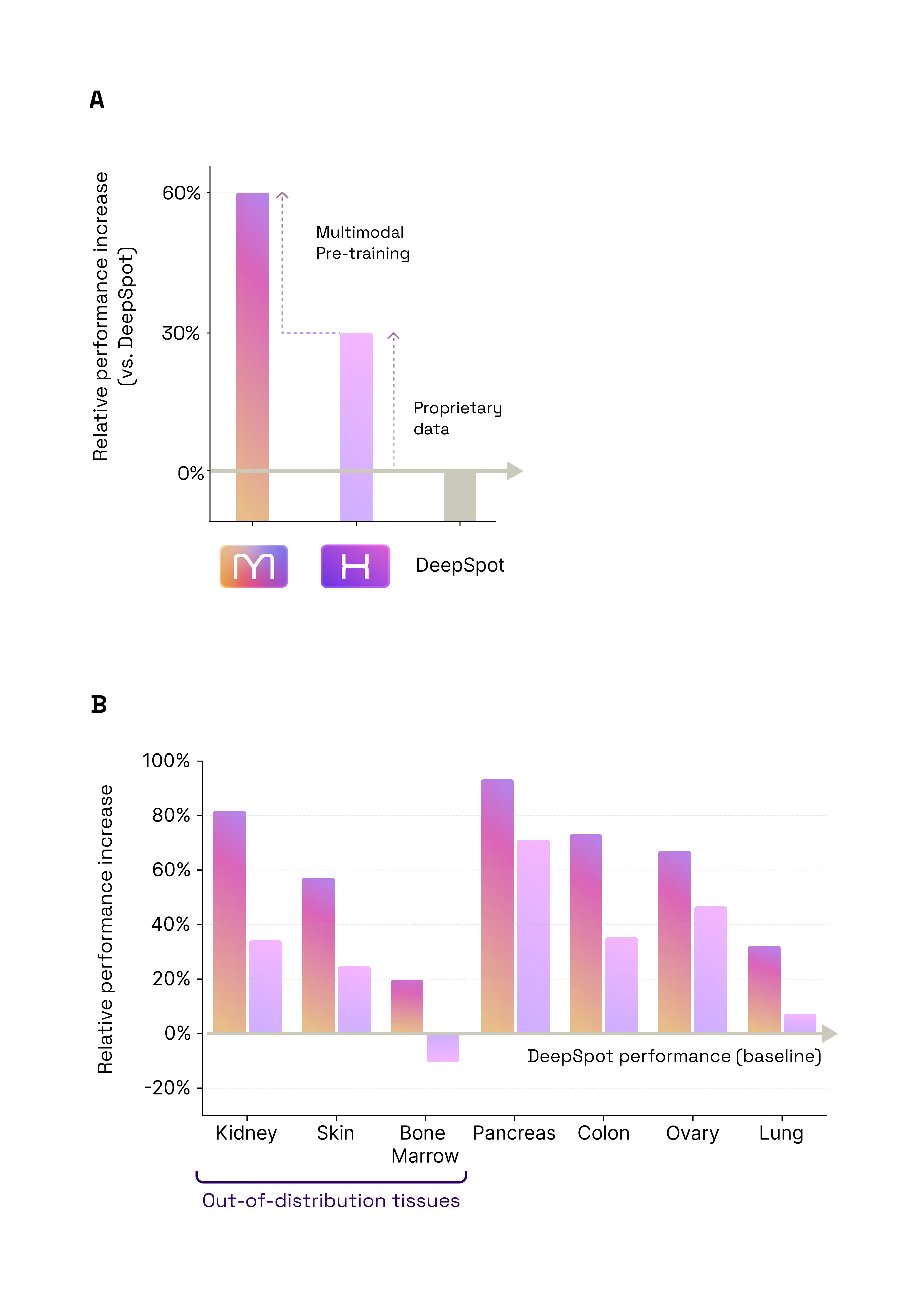
- +60% accuracy vs gold standard DeepSpot
- +30% data / +30% multimodal architecture
- No compromise on H&E encoding
- M1 outperforms gold standard H1

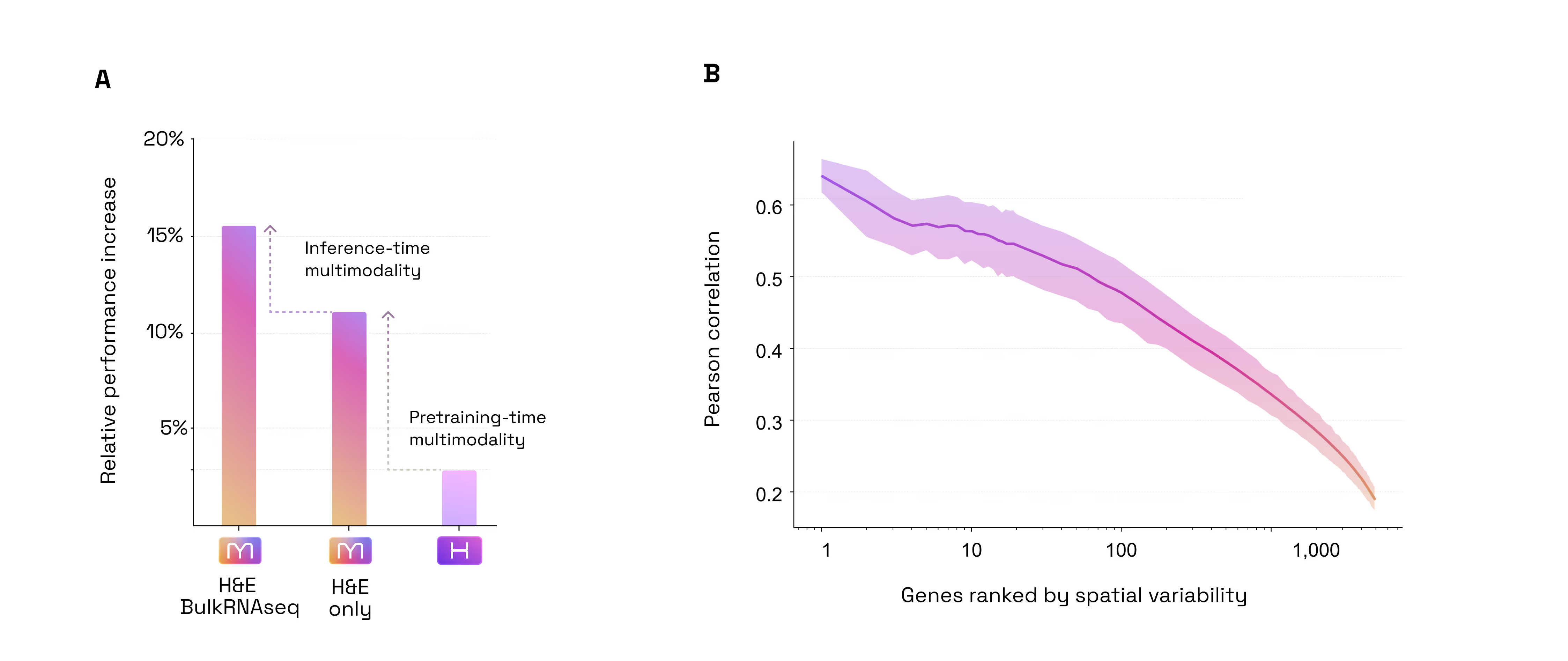
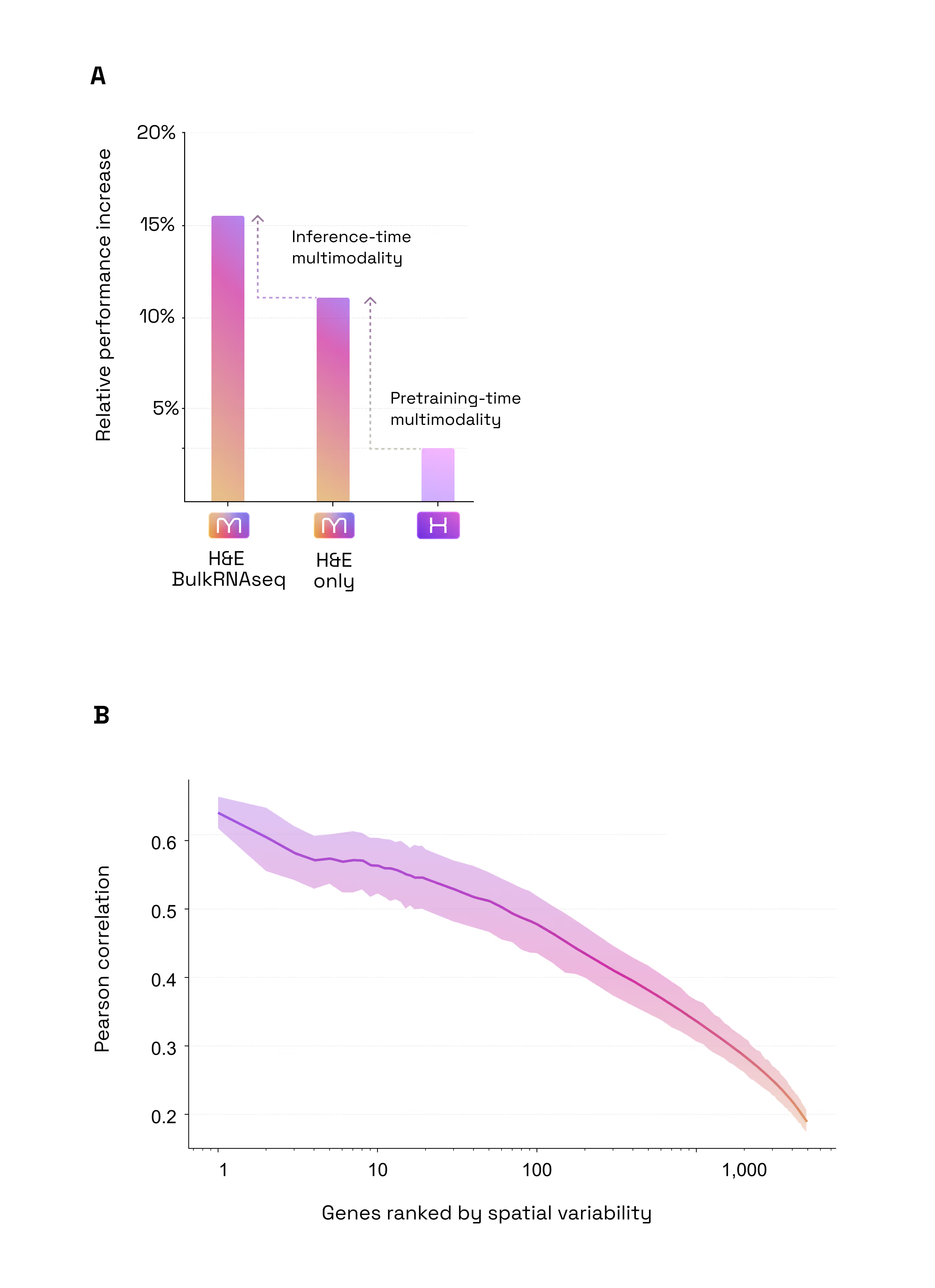
- Predict SpTx from H&E+Bulk with no re-training
- +4% accuracy boost when adding Bulk RNA-seq to H&E at inference time
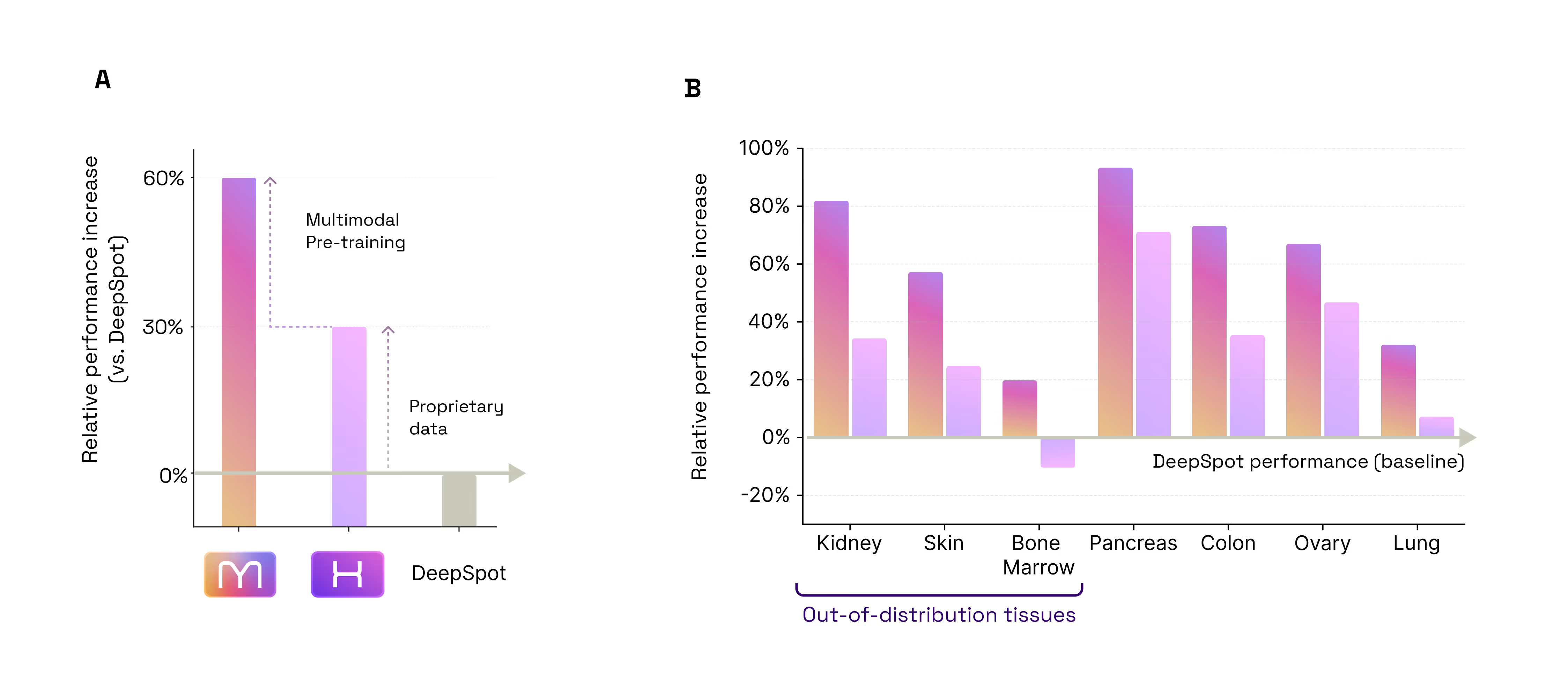
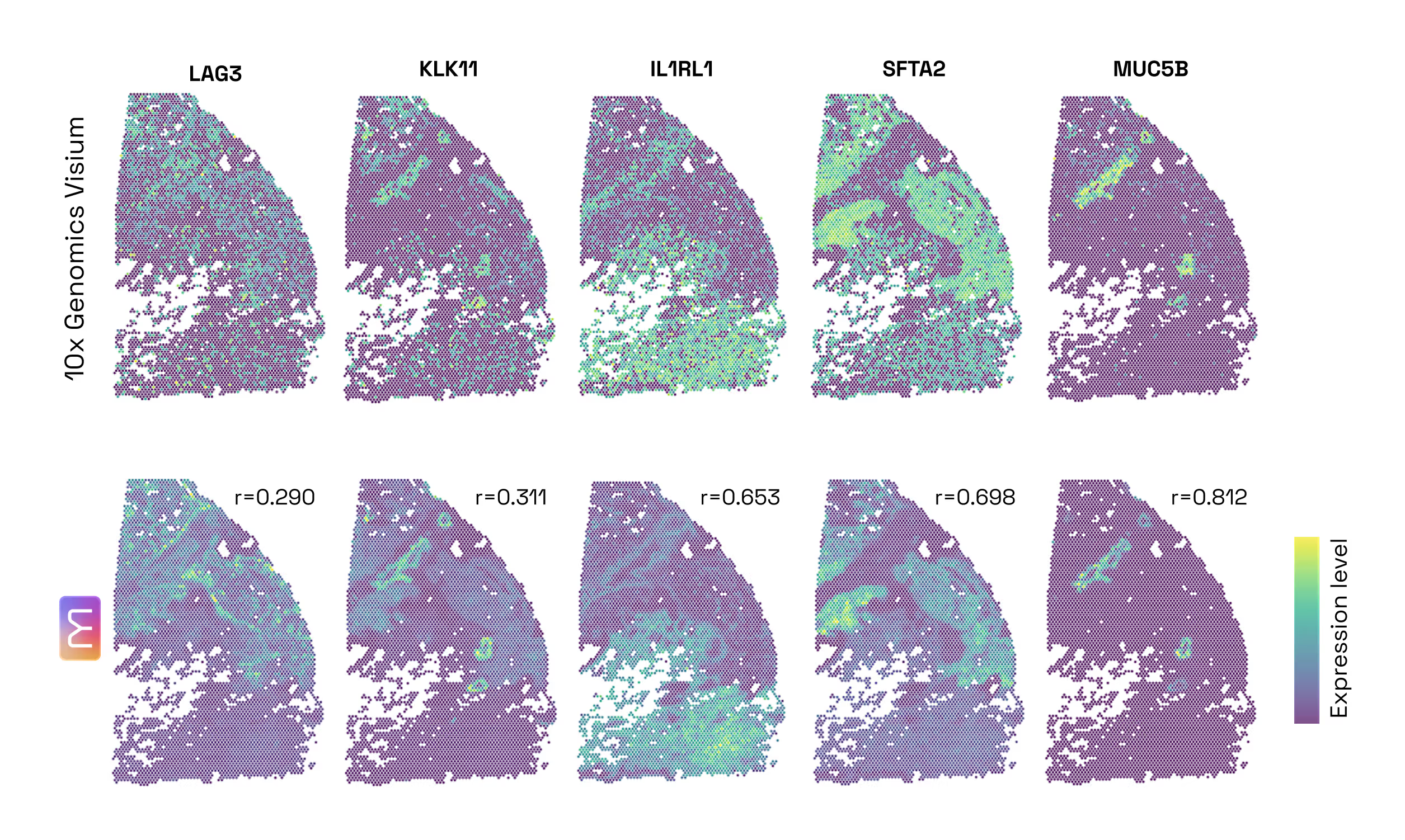
- The 60% Leap: Predicting Spatial Gene Expression from H&E. Our first test was a foundational one: Could M1 look at a standard, low-cost H&E slide and accurately predict the activity of 6,000 different genes? When compared to DeepSpot (considered one of the best models in this particular field today), M1 delivered an impressive 60% improvement in accuracy. This leap in performance comes from two sources: 30% from our massive proprietary data, and 30% from our unique multimodal architecture and training method (Figure 2.1).
- Multimodal Inference Boost: H&E + Bulk RNA-seq. Most models are limited to the data they see at the moment of inference. M1 is different, it can seamlessly ingest unimodal or multimodal data. Our second test was to assess if M1 benefits from multimodal inputs to predict spatial gene expression. We found that the model naturally "gets smarter" when it is prompted with additional modalities. By adding Bulk RNA-seq data alongside the tissue image at inference time, the model became 4% more accurate than using the image alone (Figure 4). Because it is built to benefit from every fragment of available data, M1 doesn’t require re-training to incorporate this extra context: when available, it simply uses the bulk profile as a "guide" to refine its spatial predictions. (Figure 2.2)
- Multimodal Training Preserves or Beats Specialization Encoding: A State-of-the-Art H&E Encoder. A common fear in AI is that adding complexity (like multimodality) degrades performance on simple, unimodal tasks. Our third test was to assess whether M1 is a good generic encoder for H&E images in spite of being trained on multimodal data. For that purpose, we tested M1 against H1, one of the industry's current gold standards for tissue analysis (as of May 2026, H1 is ranked as the #1 foundation model on public leaderboards such as HEST and PathBench) while being pretrained on a set of H&E images very similar to that used by M1. On the HEST benchmark (predicting gene expression), M1 outperformed H1 by ~4% (average Pearson correlation 0.440 vs 0.423). This confirms that by training on multiple modalities, M1 has developed a "sharper eye" for subtle morphological and molecular details that image-only models can’t resolve. When it comes to the bread and butter of pathology, predicting tumor subtypes, mutations, or whether a cancer has spread, M1 matched the performance of H1 (mean AUC across 9 tasks: 0.664 vs 0.661). Overall, this proves that M1 doesn't trade-off basic diagnostic power for molecular insight; it enhances the former while mastering the latter (Figure 2.3).
The Universal Language of Biology: Why a Generalist Beats the Specialists
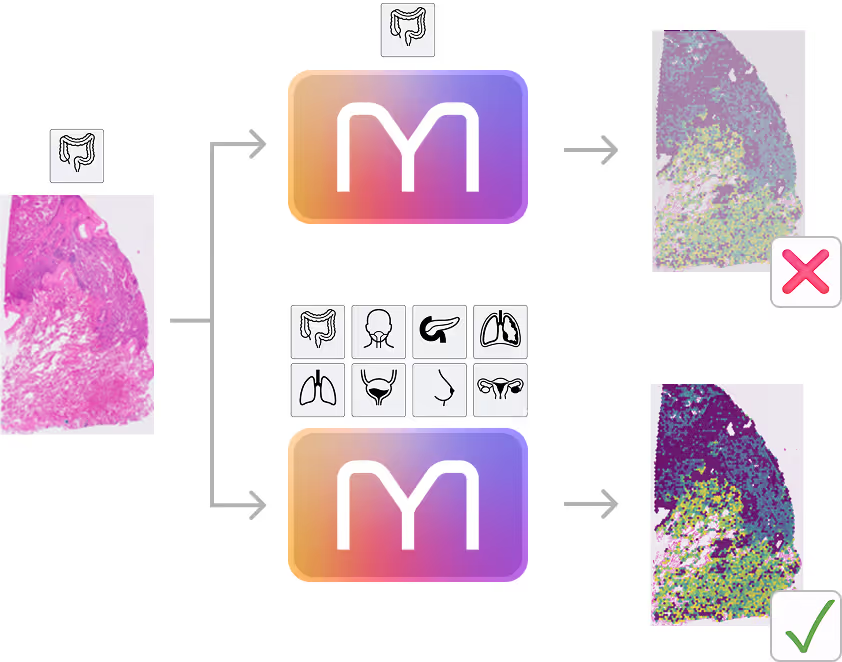
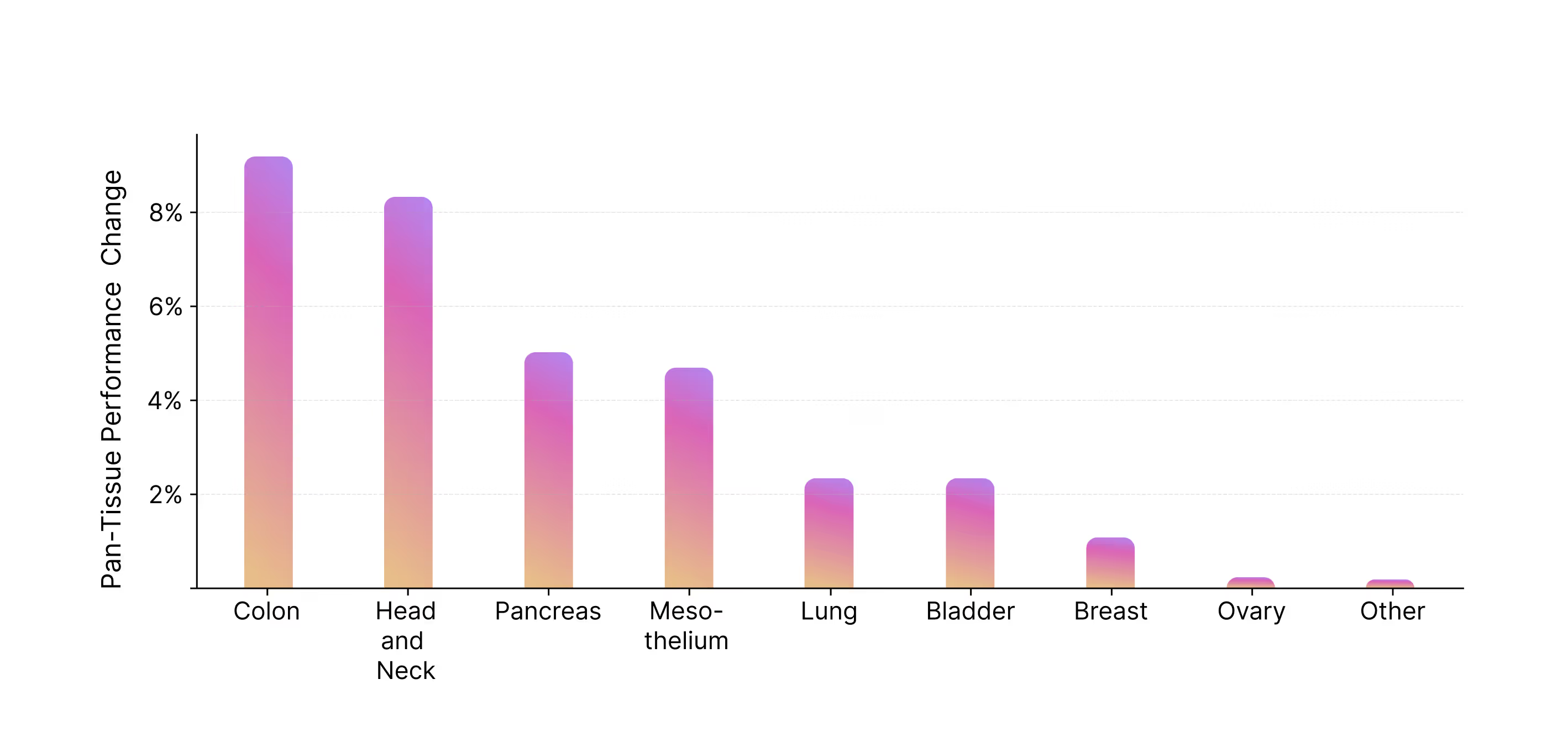
- M1 transfers knowledge between indications
- +8% on Colon and Head & Neck cancers for the generalist vs specialist model

- Zero-shot generalization capabilities
- M1 outperforms specialist models (here DeepSpot) on indications it has never seen before
Predicting biology that was previously invisible


What this means for drug discovery today
Traditional biomarkers are often unimodal—either a specific genetic mutation or a visual pathology score (like PD-L1). M1 enables the creation of multimodal digital biomarkers. By combining the structural context of H&E with the depth of Bulk RNA-seq, M1 identifies complex "signatures" of drug response that neither modality could catch alone. This leads to higher sensitivity in patient stratification and a reduced risk of trial failure.
Millions of existing pathology slides from historical trials represent a gold mine of untapped data. Many of these trials lack spatial molecular data because the technology didn't exist or tissue was too scarce. M1 allows researchers to perform retrospective molecular analysis at scale, virtually enriching old cohorts with spatial gene expression profiles. By linking these new maps to known long-term outcomes, we can discover why some patients responded years before we had the tools to understand their biology.
Precision medicine is currently bottlenecked by the cost of assays. At roughly $10 for a routine H&E slide, M1 acts as a high-fidelity first-pass filter. Instead of committing $10,000 per sample for spatial transcriptomics across an entire cohort, researchers can use M1 to screen thousands of patients and then target expensive wet-lab assays only on the most informative samples. This makes 1,000-patient spatial studies financially and logistically viable for the first time.
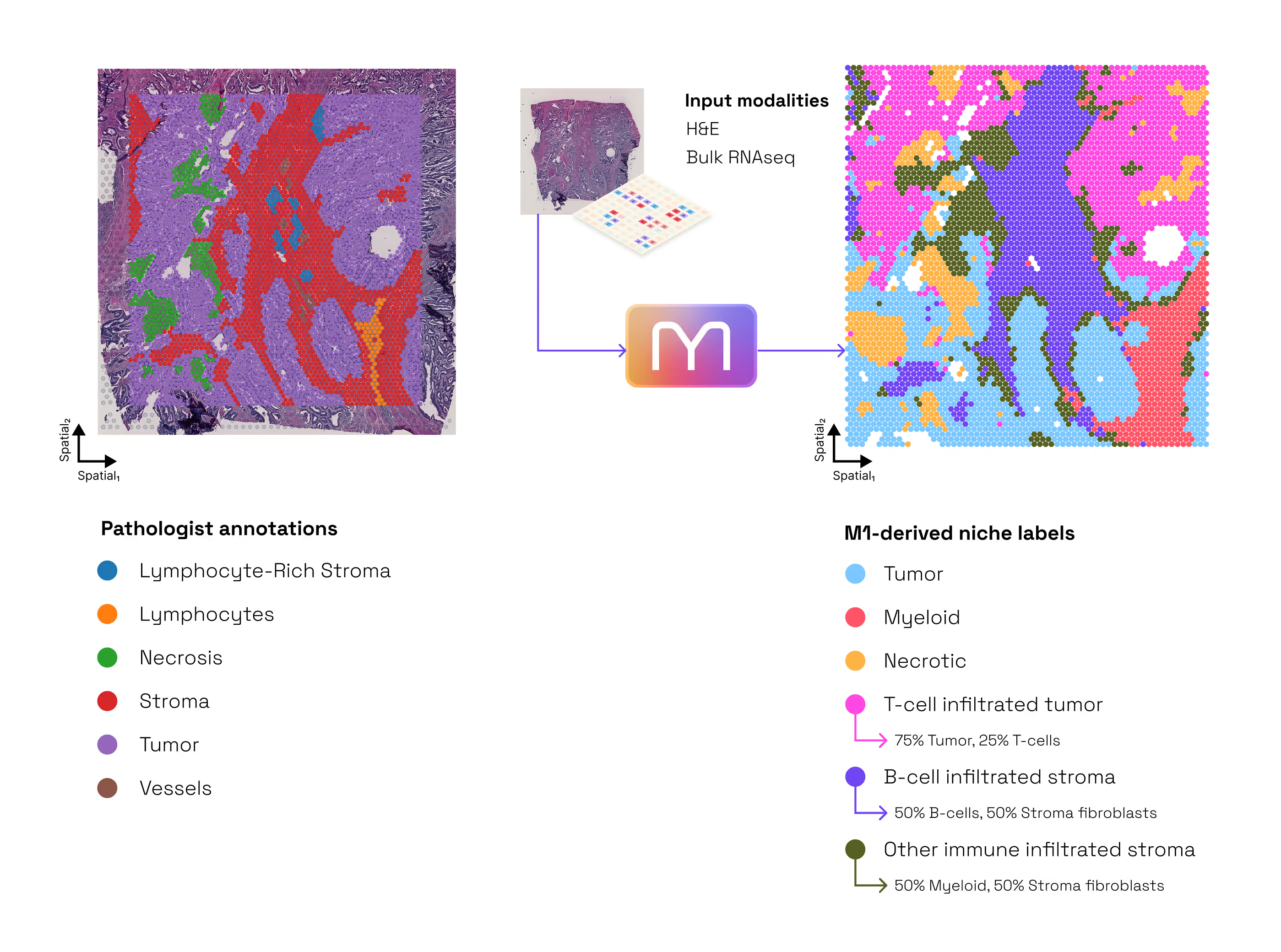
M1 allows researchers to "prime" their experiments with multimodal context. For example, by predicting spatial expression patterns across a whole-slide image first, it allows the user to target downstream micro-dissection or high-res assays to the specific regions of interest identified by the model. Additionally, M1 identifies immune substructures and metabolic niches that are invisible to the human eye, providing a "digital twin" of the tumor microenvironment without requiring additional tissue or staining costs (Figure 9 and deep dive).
Because M1 has learned a universal grammar of biology, it allows for "knowledge transfer." Insights gained from a large-scale oncology cohort can be applied to data-sparse rare diseases where tissue samples are precious. This capability is a game-changer for drug repurposing, allowing us to see if the "immune signature" of a successful therapy in one indication is present in another, underserved disease.

Performance scales with data, and has not plateaued
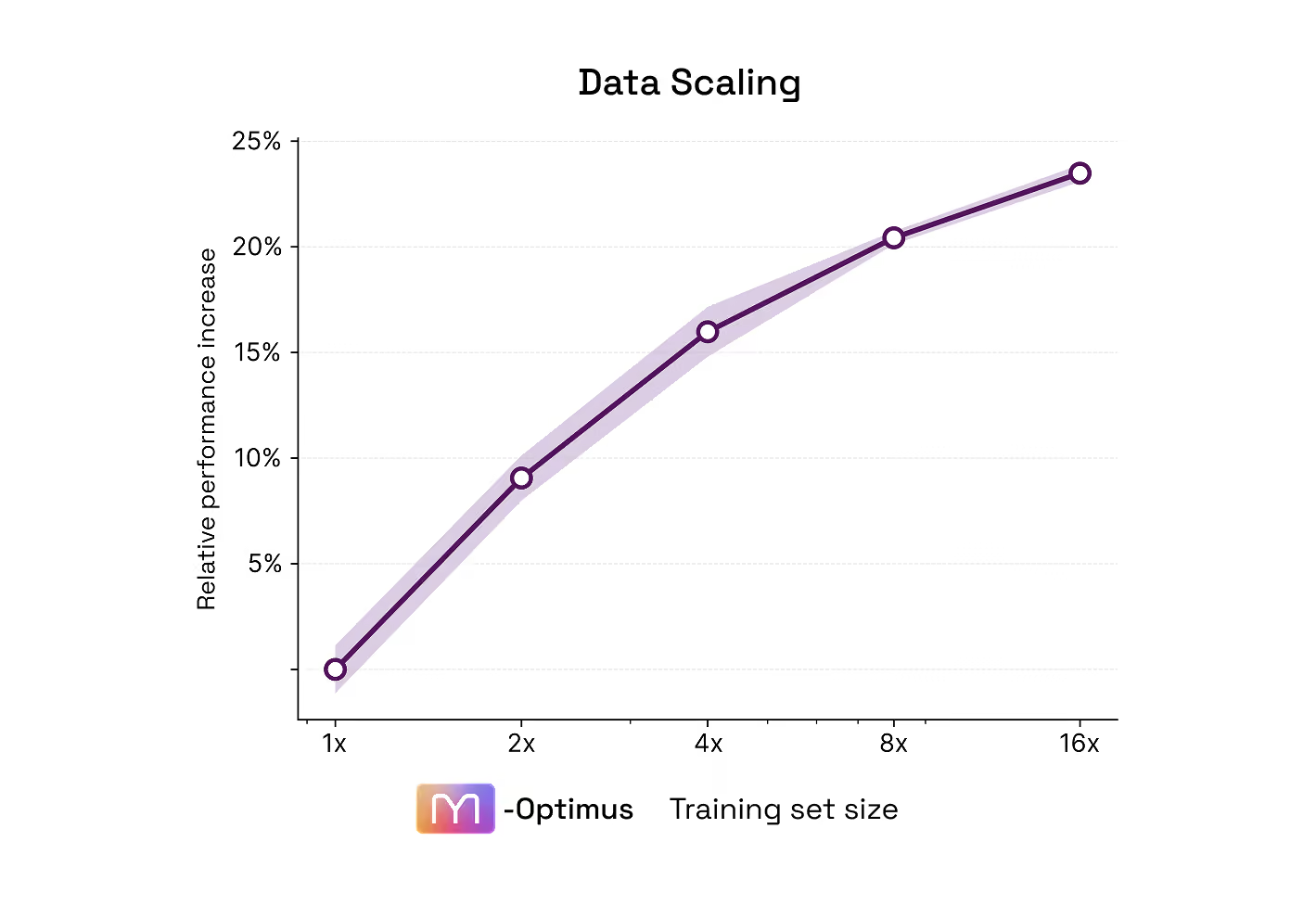
What comes next
We are now building the datasets and architecture that will enable the next iterations of M-Optimus:
Better data
Broader modalities
Higher resolution
We have barely scratched the surface. The next post in this series, exploring biological insights, will begin to show why.
Access
References
Ganguly K, Kishore U, Metkari SM and Madan T. Immunomodulatory Role of Surfactant Protein-D in a Transgenic Adenocarcinoma of Mouse Prostate (TRAMP) Model. Front Immunol 13:930449, 2022.
Jaume, G., Doucet, P., Song, A. et al. HEST-1k: A Dataset For Spatial Transcriptomics and Histology Image Analysis. Advances in Neural Information Processing Systems 37:53798–53833, 2024.
Mohammadi, A., Inayatullah, M., Schlosser, A. et al. Pulmonary surfactant protein D reduces lung cancer progression associated with decreased IL-4/STAT6 signaling. npj Precis Onc 10:164, 2026.
Mouillet-Richard, S., Cazelles, A., Pilati, C. et al. Profiling colon cancer architecture with spatial transcriptomics identifies clinically relevant stromal ecotypes, Preprint (Version 1) available at Research Square, 2026.
Nonchev, K., Dawo, S., Silina, K. et al. DeepSpot: Leveraging Spatial Context for Enhanced Spatial Transcriptomics Prediction from H&E Images. Preprint medRxiv 2025.02.09.25321567, 2025.
Schmauch, B., Herpin, L., Olivier, A. et al. A deep learning-based multiscale integration of spatial omics with tumor morphology. Nat Commun 16:11674, 2025.
Umeda Y, Hasegawa Y, Otsuka M, et al. Surfactant protein D inhibits activation of non-small cell lung cancer-associated mutant EGFR and affects clinical outcomes of patients. Oncogene 36(46):6432-6445, 2017.
Wang, C., Chan, A.S., Fu, X. et al. Benchmarking the translational potential of spatial gene expression prediction from histology. Nat Commun 16:1544, 2025.